Ursache

Was ist Morbus Pompe?
Morbus Pompe (auch als Pompe-Krankheit, Pompe Disease bezeichnet) ist eine angeborene lysosomale Speicherkrankheit (Glykogenspeicherkrankheit Typ II). Aufgrund eines Defekts des Glykogen-abbauenden Enzyms saure ɑ-Glucosidase (GAA) wird Glykogen – eine wichtige Speicherform der Kohlenhydrate – in den „Verdauungsorganen“ der Zellen, den Lysosomen, nicht ausreichend verstoffwechselt. Mit der Zeit häuft sich Glykogen innerhalb der Lysosomen in den Zellen verschiedener Geweben und Organen des Körpers an und schädigt diese.

Morbus Pompe wird durch Mutationen im GAA-Gen verursacht

Diese Mutationen können dazu führen, dass das Enzym saure ɑ-Glucosidase fehlt oder nicht richtig funktioniert

Wenn GAA fehlt oder nicht richtig funktioniert, reichert sich Glykogen in den Lysosomen der Zellen an

Es kommt zu Zellschäden in den betroffenen Geweben, die Ursache mannigfaltiger Beschwerden bei Morbus Pompe sind
Mechanismus der übermäßigen Glykogen-Anreicherung in den Lysosomen
Da Muskelzellen (neben der Leber) den wichtigsten Speicherort für Glykogen darstellen, sind sie besonders stark von der Glykogen-Anreicherung betroffen. Dies äußert sich je nach Krankheitsverlauf in einem zunehmenden und unwiederbringlichen Funktionsverlust der rumpfnahen Skelettmuskulatur sowie der Atemmuskulatur. Aus diesem Grund wird Morbus Pompe auch den stoffwechselbedingten Muskelerkrankungen (metabolischen Myopathien) zugeordnet.
In schweren Fällen kann auch das Herz betroffen sein.
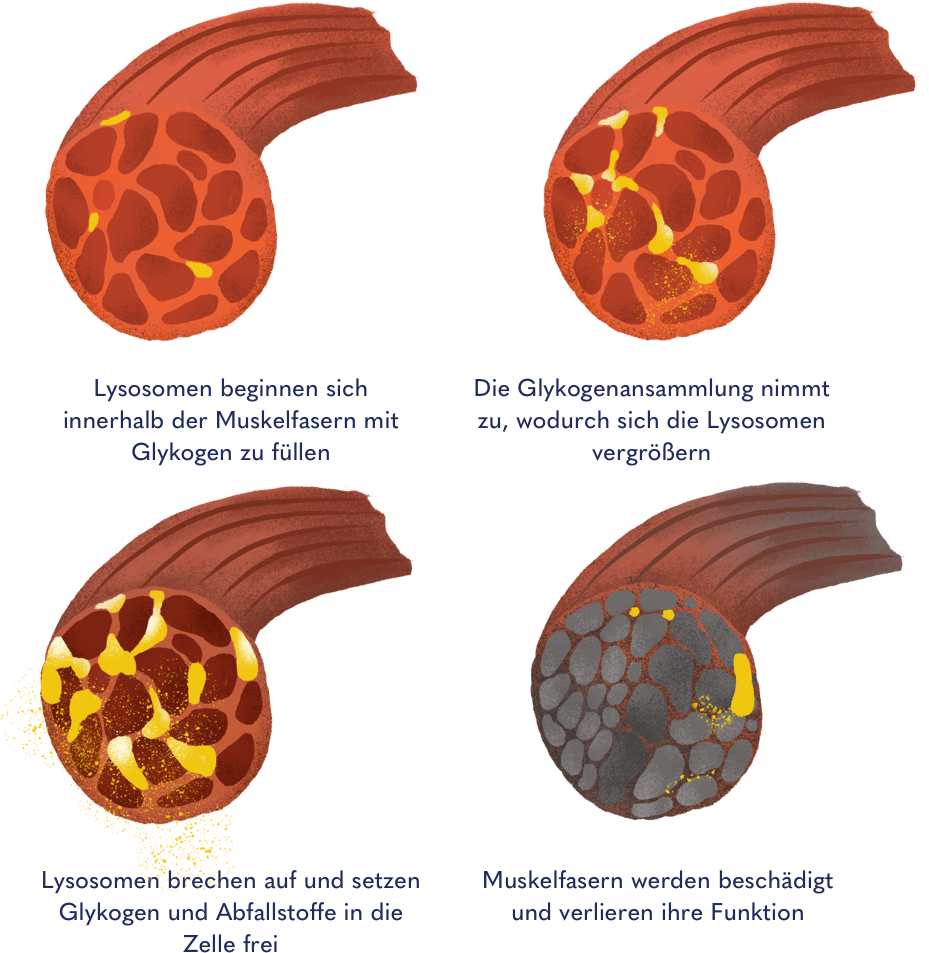
Zunehmende Schädigung der Muskelfasern durch Anreicherung von Glykogen
Wer erkrankt an Morbus Pompe?
Morbus Pompe gilt mit etwa 5,3 Fällen je 100.000 Lebendgeburten als seltene Erkrankung. Die zugrundeliegende Mutation im GAA-Gen kann von gesunden Eltern, die beide lediglich Träger der Mutation sind, an ihre Nachkommen weitergegeben werden. Nur wenn die Kinder zwei mutierte Allele – eines von der Mutter und eines vom Vater – erhalten, tritt die Erkrankung auf. Die Wahrscheinlichkeit dafür beträgt 25 %. Kinder, die nur eine mutierte Version des GAA-Gens von Mutter oder Vater erhalten haben (50 %), sind heterozygot und erkranken nicht. Allerdings können sie als Anlageträger das defekte Gen wiederum an ihre Nachkommen weiter vererben. Wenn ein Elternteil erkrankt ist und ein Elternteil Träger der GAA-Mutation ist, liegt die Wahrscheinlichkeit, dass auch ihre Kinder an Morbus Pompe erkranken, bei 50 %.
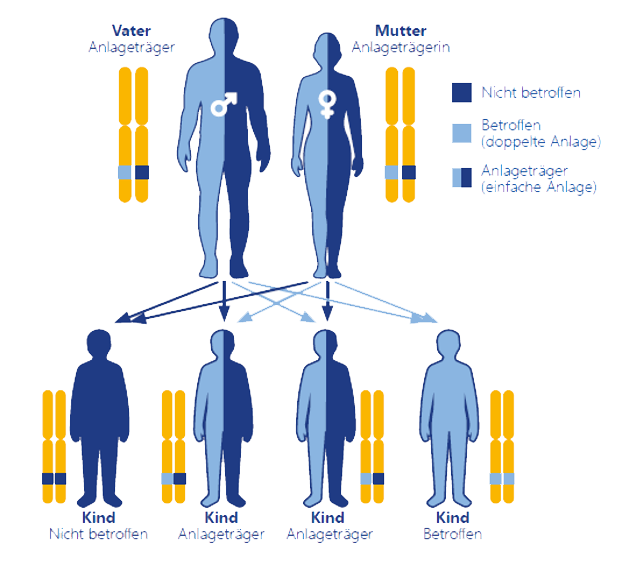
Autosomal-rezessive Vererbung von Morbus Pompe
Bislang sind mehr als 600 krankheitsverursachende Varianten des GAA-Gens identifiziert worden. Je nach vererbter Genvariante wird weniger GAA gebildet als bei Gesunden (quantitativer Defekt) oder die Funktion des GAA-Enzyms ist beeinträchtigt (qualitativer Defekt). Dies erklärt mitunter auch die Unterschiede hinsichtlich des Erkrankungsbeginns und der Ausprägung der Symptome.

Es lassen sich zwei Verlaufsformen der Pompe-Krankheit unterscheiden – die Infantile Form des Morbus Pompe (Infantile-Onset Pompe Disease, IOPD), die schon früh im ersten Lebensjahr auftritt, sowie die Späte Verlaufsform des Morbus Pompe (Late-Onset Pompe Disease, LOPD), die sich (teilweise erst Jahre bis Jahrzehnte) nach dem ersten Lebensjahr manifestiert.
In der Regel besteht ein Zusammenhang zwischen der verbleibenden Enzymaktivität und dem Schweregrad der klinischen Symptome – eine sogenannte Genotyp-Phänotyp-Korrelation. Betroffene mit der infantilen Form weisen meist zwei GAA-Mutationen auf, die einen nahezu vollständigen Verlust der Enzymaktivität zur Folge haben. Das Vorhandensein mindestens einer “milden” Mutation führt hingegen eher zur Ausprägung der weniger schweren späten Verlaufsform (LOPD).